

图:会议材料截图
会议首先针对非临床审评的大方向进行了分别阐述,描述了药物的作用机制研究,药效学,药代动力学以及安全性评价的重要性。
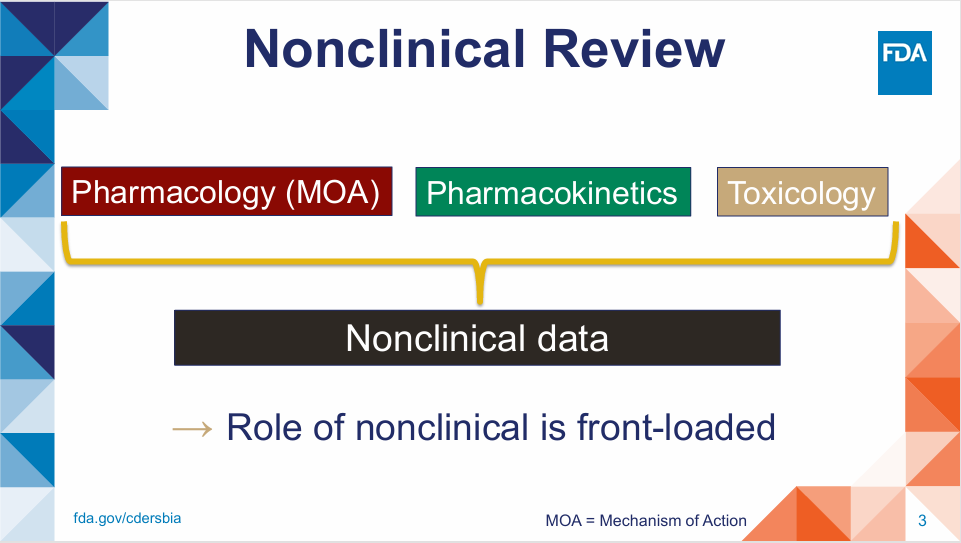
图:会议材料截图
其次对于抗肿瘤药物的开发,CDER建议参考ICH S9《抗肿瘤药物非临床评价指导原则》(https://www.fda.gov/media/73161/download),《抗肿瘤药物非临床评价指导原则问答》(https://www.fda.gov/media/100344/download)和 S6(R1)《生物制品的临床前安全性评价》(https://www.fda.gov/media/72028/download)。特别地,对于小分子化药而言,建议在两个动物种属上完成28天的GLP毒理实验,而生物制品,只要能阐明其科学性和合理性,在一个相关动物种属上完成GLP毒理实验也是可以被接受的。同时,在减少申办方非临床毒理报告的准备时间上,FDA也做出了一些让步,会议中提到在IND阶段可以选择递交含有已签署认证的组织病理学报告的毒理学报告草稿。

图:会议材料截图

图:会议材料截图
对于具有免疫激动特性的生物制品,ICH S9 强调应考虑使用最小预期生物效应水平(MABEL)来选择起始剂量。会议中推荐了一些DHOT发表的关于FIH起始剂量选择的文章,可以提供参考:
• An FDA oncology analysis of immune activating products and first-in-human dose selection. PMID:27743776. (https://pubmed.ncbi.nlm.nih.gov/27743776/)
• An FDA oncology analysis of CD3 bispecific constructs and first-in-human dose selection. PMID: 28887049. (https://pubmed.ncbi.nlm.nih.gov/28887049/)
• An FDA oncology analysis of antibody-drug conjugates. PMlD:25661711. (https://pubmed.ncbi.nlm.nih.gov/25661711/)
• An FDA oncology analysis of toxicities associated with PBD-containing antibody-drug conjugates. PMID: 31325532. (https://pubmed.ncbi.nlm.nih.gov/31325532/)
• Pharmacokinetic models for first-in-human dose selection of immune-activating products in oncology. PMID: 38561147 (https://pubmed.ncbi.nlm.nih.gov/38561147/)

图:会议材料截图

图:会议材料截图
会议中着重强调了安全性研究(主要是毒理学研究)的GLP合规性的重要性,以及毒理学研究中包含的试验终点。
会议表示,GLP 是一种质量体系,对非临床研究的设计、执行、监控、记录、存档和报告的条件进行规范,具体地描述可以见21 CFR part 58 (https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=58)。而在申办方进行GLP试验时,一定要提前与CRO或实验室进行确认,需要确保他们能提供非临床数据交换标准数据(SEND),2017 年 12 月 17 日之后启动的商业IND,单剂量毒性、重复剂量毒性和致癌性研究需要提供SEND 数据。2020 年 3 月 15 日之后启动的商业IND,还需要提交心血管和呼吸安全药理学试验地SEND数据。如果对GLP试验设计存在疑问,可以先咨询专家,或者递交pre-IND 会议申请给FDA。其他需要在首次人体实验(FIH)之前完成的包括:MOA研究,特异性研究,浓度响应研究,安全药理研究(hERG和体内试验)对于免疫调节剂,需补充体外细胞因子释放试验。
 图:会议材料截图
图:会议材料截图
图:会议材料截图

图:会议材料截图
此外,针对GLP 毒理学研究的关键终点,FDA提到应包括:死亡率、血液学、临床观察、临床化学、体重、大体病理学、食物消耗量、器官重量、心电图(非啮齿类动物)、组织病理学、眼科和毒代动力学。具体终点可以根据需要进行增加(如细胞因子)。会议表示可参考The CFSAN Redbook (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-and-other-stakeholders-redbook-2000#TOC)对毒理学研究设计进行更进一步的了解,其内容描述了毒理学研究中应包括的研究类型(从基因到慢性毒性研究)。
对于抗肿瘤药IND,会议中提到一些需要申办方注意的问题,首先是非临床方面最常见的被Hold的理由,主要包括三点,一是FIH起始剂量和/或剂量爬坡的设计不合理,二是缺少试验或支持性数据,最后是存在严重毒性。(更多信息可参考:An FDA analysis of clinical hold deficiencies affecting investigational new drug applications for oncology products. PMID 31678263.)(https://pubmed.ncbi.nlm.nih.gov/31678263/) 其次,对于临床试验起始剂量的计算,会议也进行了强调以及指南推荐。针对小分子药物或ADC药物的推荐按照 BSA(mg/m2)计算起始剂量;对于生物制剂,按体重(mg/kg)进行计算。(BSA 换算请参照https://www.fda.gov/media/72309/download,表 3)。会议最后强调了应为生物制品/寡核苷酸的药理相关物种的选择提供数据支持。
对于特定产品CDER也推荐了相应的开发指南供申办方参考:
• 放射性药物:https://www.fda.gov/media/129547/download
• ADC:https://www.fda.gov/media/100344/download
• 新型辅料:https://www.fda.gov/media/72260/download
• 植物药:https://www.fda.gov/media/93113/download
最后,CDER推荐申办方通过申请INTERACT或者pre-IND会议及时与FDA沟通,进行早期开发的指导。会议相关要求指南请见:https://www.fda.gov/media/172311/download
撰稿人:Emma Xie