
一、引言与背景
1.1 指南适用范围
指南主要适用于依据《Federal Food, Drug, and Cosmetic Act》第 505 条提交审批的特定药品活性成分(包括任何临床给药途径)。本指南主要针对首次人体(FIH)试验中,用于健康受试者的新型小分子活性成分的检测。本指南不适用于以下情形:1. 拟依据《Public Health Service Act》第 351 条申请许可的生物制品;2. 用于治疗晚期癌症的药品活性成分;3. 具有DNA 反应性(致突变性)杂质(参见 ICH M7(R2) 指南)。
1.2 遗传毒性试验的重要性
遗传毒性试验用于识别可能引起DNA损伤的化合物,这种损伤可能导致基因突变或染色体结构异常,进而增加癌症风险。ICH M3(R2)、S2(R1)和S9等国际指南已明确了遗传毒性试验的标准组合及其在药物开发中的时机。然而,现有指南未专门针对Ames阳性药物提供后续检测的具体路径。
1.3 Ames阳性的意义与挑战
Ames试验阳性通常意味着药物具有DNA反应性,通常情况下,Ames 试验呈阳性的化合物不会继续开发,除非其用于治疗严重或危及生命的疾病,如癌症。在健康受试者中暴露于 Ames 阳性活性成分,即使单次给药,受试者癌症风险升高的概率亦不为零。因此,为支持在健康受试者中进行首次人体临床试验,应进行充分的后续测试,以进一步评估其致突变潜能。例如,若所有后续试验(如体外哺乳动物细胞突变试验或体内基因突变试验)结果均为阴性,则可依据权重证据(Weight of Evidence, WoE)方法,得出该化合物在人类中不存在遗传毒性风险的结论。
二、推荐的Ames数据分析方法以及Ames阳性活性成分或代谢物的后续检测路径
2.1 后续检测前Ames试验结果的评估
在进行后续检测前,必须对Ames试验结果进行全面评估,可参照图1的决策树,包括:• 使用OECD TG 471标准方法进行试验;
• 采用Levy等人(2019)提出的标准进行结果评估;
还需对艾姆斯试验阳性结果开展潜在影响因素评估:可通过交叉参照法,将引发Ames阳性结果的分子官能团与具有相似官能团且已掌握致癌性数据的化学物质进行比较;评估是否存在细菌特异性阳性反应或非致突变性阳性反应;若可获得人体数据,啮齿类动物与人类代谢谱的比对将具有重要参考价值,若阳性结果与人类不相关的啮齿类特异性代谢途径相关联,则开展临床试验的风险关注度将相应降低。
2.2 体外小鼠淋巴瘤试验(MLA)和哺乳动物细胞HPRT试验
后续检测实验最先进行以下任一试验:• 小鼠淋巴瘤试验(MLA):可检测基因突变、染色体断裂和非整倍体,试验应遵循OECD 2016a指南,MLA必须进行集落大小分析, 大集落突变体将是与Ames阳性发现一致的致突变事件的证据;
• 哺乳动物细胞HPRT试验:主要用于检测基因点突变或小片段缺失。
2.3 体内基因突变试验
若体外哺乳动物细胞试验为阴性,则需进行体内突变试验,以评估在更接近人体的生理条件下的致突变潜力。推荐方法包括:• TGR 基因突变试验可评估多组织的体内致突变性(Lambert et al. 2005; Nohmi et al. 2017; OECD 2022b)。加入 Pig-a 终点(OECD 2022a)可提高效率:若结果为阳性,可免除组织分析;若为阴性,则应进行 TGR 组织突变分析。
• 在有充分依据的情况下,仅进行 28 天 Pig-a 试验也可接受无需开展TGR突变研究,例如基于 ADME 与一般毒理学数据(例如组织病理学、骨髓涂片评估结果提示组织选择性反应可能性低、血浆中活性成分暴露量高而组织暴露量低等)。
• OECD TG 488 (2022b) 和470 (2022a) 对TGR试验与Pig-a试验的规范实施提供了详细技术指导。TGR 组织选择应由给药途径、ADME 和毒性数据指导,并分析多个与暴露相关的组织以增强证据权重。阴性结果需经生物分析方法验证暴露水平支持。
• 微核或彗星试验主要检测染色体断裂,不能充分反映Ames阳性的活性成分引起的小规模基因突变,因此不适用于评估Ames阳性结果(Robison et al. 2021)。
2.4 活性成分的代谢物呈Ames阳性的后续检测的考虑
• 若代谢物在Ames试验中呈阳性,应参照上述药物活性的评估流程进行体外和体内检测,在某些情况下,测试分离的代谢物可能是有利的。• 对于在人类中出现的不同比例或新的代谢产物,应按照 ICH M7(R2) 的规定进行处理。
• 若代谢物浓度极低(如在毒理学关注阈值水平),则可参照ICH M7(R2)对遗传毒性杂质的处理方式,无需进行上述广泛的体外和体内遗传毒性后续检测。
2.5 致癌性试验的必要性
当作为证据权重评估的一部分考虑时,体外和体内突变试验均为阴性的结果可能有助于为在健康人类受试者中进行 FIH 临床试验提供充分的安全性评估,但仍不能完全排除致癌风险。在 FIH 试验中招募健康受试者应予以论证(即,为什么给健康受试者施用含有相关活性成分的研究性新药而不是患者,知情同意文件中是否有足够信息描述致突变潜力的发现,以便受试者了解潜在风险)。然而,在大多数情况下,仍预期在新药申请提交时或之前进行啮齿类动物致癌性研究,并且如果需要,此类研究可能在 IND 药物开发阶段更早进行。对于在体外哺乳动物试验和/或体内突变试验中也呈阳性的Ames阳性活性成分,可考虑进行致癌性研究,以确定阳性信号是否转化为肿瘤发现。在这种情况下,需要阴性的致癌性研究来支持在健康受试者中进行 FIH 试验。
三、对Ames阳性活性成分或代谢物进行证据权重评估的考虑因素
3.1 结构-活性关系分析
应通过 QSAR 和交叉参照法(read-across) 评估化合物结构的潜在致突变性结构警报。与已知致突变/致癌化合物结构相似的数据可增加风险,而与阴性对照化合物相似的数据可降低风险。
3.2 代谢特征比较
应评估啮齿类与人类的代谢谱。若体外阳性结果与仅在人类无关的啮齿类特异性代谢物相关,可降低关注;若在人类中存在潜在致突变代谢物,则增加关注。若代谢数据表明致突变形式在体内迅速失活,也可降低关注。必要时可使用human S9 进行补充评估,但其结果仅作证据权重的一部分。
3.3 关键排除标准
以下情况不支持在健康受试者中进行FIH试验:1. 活性成分结构类似已知致癌物(如 N-亚硝基化合物、多环芳烃等);
2. MLA或HPRT试验阳性或结果不明确,若在标准遗传毒性试验组合的其他检测中(如体外染色体畸变试验、体内微核试验)出现阳性结果,将进一步佐证不应在健康受试者中开展试验。唯一可行的路径可能是在疾病患者群体中开展首次人体试验——前提是存在可接受的风险获益比,或者替代性地进行6个月rasH2小鼠或2年大鼠/小鼠致癌性研究。若选择开展此类研究,必须获得阴性结果方可继续开发进程并在健康受试者中实施首次人体试验;
3. 虽 MLA/HPRT 阴性,但 TGR 或 Pig-a 试验阳性的Ames阳性活性成分。唯一可行的路径可能是在疾病患者群体中开展首次人体试验——前提是存在可接受的风险获益比,或者替代性地进行6个月rasH2小鼠或2年大鼠/小鼠致癌性研究。若选择开展此类研究,必须获得阴性结果方可继续开发进程并在健康受试者中实施首次人体试验。
3.4 极少数情况
在大多数情况下,基于后续检测的评估结果(如体内或体外致突变性试验呈阳性),含有 Ames 阳性活性成分的药物通常不适合在健康受试者中开展首次人体(FIH)试验;但在极少数情况下,若所获得的安全性证据(例如体外 MLA 或 HPRT 试验结果为阴性,且体内突变试验亦为阴性)或其他风险减轻因素具有足够说服力,则可能得出以下结论:在健康受试者中使用该 Ames 阳性成分进行 FIH 试验是合理安全的(参见决策树方框 10b)。需要强调的是,即使完成上述推荐的后续检测,也无法完全排除 Ames 阳性结果所带来的潜在风险,仍存在一定程度的残留不确定性。因此,若缺乏充分的风险减轻依据,Ames 阳性活性成分的开发应仅限于针对严重或危及生命、且存在未满足医疗需求的疾病领域。
四、与CDER的早期沟通
FDA强烈建议申办者在提交涉及Ames阳性药物的IND前,通过pre-IND会议与CDER审评部门进行沟通,就后续检测策略和证据权重评估达成共识。
五、总结与图示
指南附有图1:决策树,清晰展示了从Ames试验阳性出发,经体外和体内检测,最终判断是否适合在健康受试者中进行FIH试验的全过程。该图强调:• 必须同时满足体外哺乳动物细胞突变试验阴性和体内基因突变试验阴性;
• 任一环节阳性即排除在健康受试者中试验的可能;
• 最终决定应基于整体证据权重,并充分考虑残留风险。
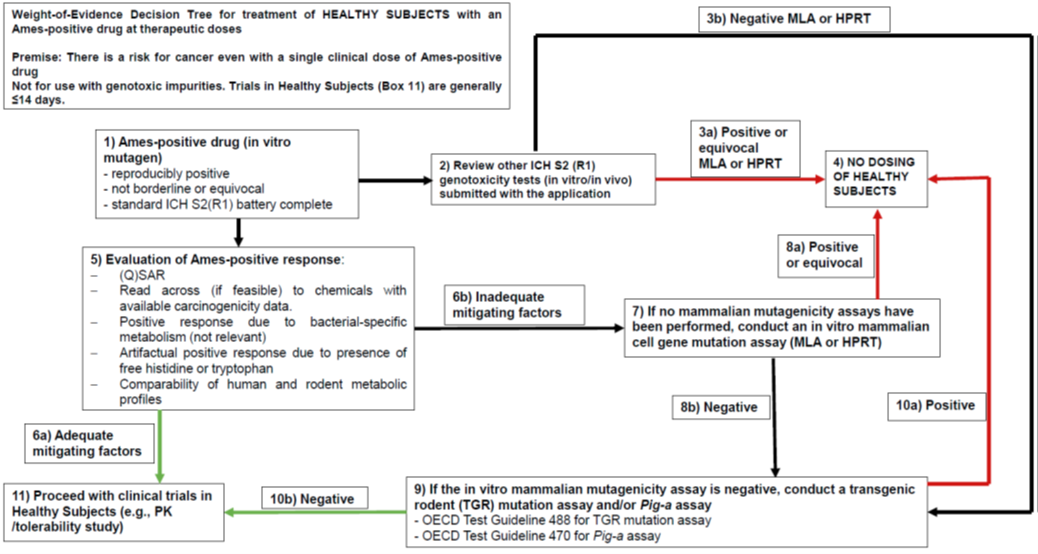
图 1:Ames阳性药物(活性成分)后续检测决策树
撰稿人:Zoey Pang, Grace Tian