


图:RMAT和BT的申请和认定数量趋势
来源:CMC Development and Readiness Pilot (CDRP) Program, Regulatory Education for Industry (REdI)-2023 Annual Conference, June 8-9, 2023
为了加快药品开发进程,临床研究方面采取了一些加速措施,包括对临床试验设计的调整、用2期临床数据支持产品上市、临床疗效替代终点等。但是对CMC方面的要求却没有任何变化。显然,CMC成为了产品加快开发的限速点。为了使CMC跟上药品开发的步伐,FDA提出了CDRP计划,一方面督促开发者尽早将满足上市条件的CMC研究提上日程,另一方面也为开发者提供了更多和FDA沟通交流的机会。
CDRP优点
鼓励申请方尽早与FDA沟通,解决CMC开发中遇到的问题。比如:生产、纯化、检测、生物信息学、稳定性研究、方法验证等问题
除了法规中规定的正式会议,额外提供两次CMC专题会(B类会议形式)
两次CMC专题会后的后续交流机会(解决会议中的遗留问题或者衍生问题)
FDA将总结该计划的项目经验作为案例向公众分享,使更多相关领域的研究者学习获益
CDRP符合性条件
产品已获批临床试验申请(Investigational New Drug Application, IND),且该IND是激活状态
IND是以eCTD形式递交
• FDA豁免了eCTD形式递交的IND除外
获批的IND需是2期临床结束之前的临床研究,以下情况例外
• 临床开发遵从创新试验设计,比如复杂创新试验设计计划(Complex Innovative Trial Design Program, CID) 参与者符合条件。CID计划是与临床试验设计相关的讨论,重点关注药物后期开发,而非首次用于人体试验。更多关于CID的信息详见FDA官网https://www.fda.gov/drugs/development-resources/complex-innovative-trial-design-meeting-program
• 该药物用于治疗罕见病
承诺进行加速临床开发的同时执行CMC研究计划
组合产品(combination product)
• 过于复杂的组合产品可能不会被列入CDRP计划中
须有RMAT或BT认定资格
• 如果向CBER递交CDRP申请,必须先有RMAT或BT资格认定才能申请CDRP
• 如果向CDER递交CDRP申请,RMAT或BT可与CDRP同步申请
CDRP申请材料
向CBER或CDER递交CDRP的书面申请
• 以IND的补充申请形式递交(IND amendment)
IND号以及产品加速开发的资格认定
• BT或RMAT获得资格认定的日期
如该产品下有其它活跃的IND申请,说明这些IND的进展状态
如果申请方有其它已上市产品,提供这些产品的信息、上市号
• 包括除美国之外在其他国家上市的产品
• 描述申请方的产品生产经验水平(生产类似产品或不同类型产品)
• 如果委托CMO生产,说明该CMO的生产经验水平
提供CMC研究计划
• 当前CMC开发进展,包括IND内容以外的正在进行的活动,比如计划更换CMO、生产场地
• 已有的产品特性研究结果以及初步判定的关键质量属性(critical quality attributes, COA)
• 当前的DS、DP生产过程和控制策略(包括鉴定和开发检测方法),上市生产的计划和控制策略(包括必要的微生物控制策略)
• 如何建立完整的试剂供应链
• 工艺验证总体计划
• 结合临床加速开发时间窗,制定生产准备计划,突出潜在风险
说明该IND是否因为CMC的原因被临床叫停(clinical hold),如果是,说明hold的状态
和FDA开会的计划和大概的时间
CDRP入选标准
申请方可以随时向FDA递交申请,没有时间规定,FDA对申请材料会进行滚动审阅,在申请后的180天内给予回复。评判要素如下
• 患者能更早获益
• 产品的新颖性
• 产品或其制造工艺(包括技术)的复杂性
• 申请方的总体生产经验(如果申请方已有相关领域的生产经验,那么不太可能会入选,因为FDA认为该申请方不需要获取FDA更多的指导意见)
信息披露协议(Disclosure Agreement)
上述提到FDA会从CDRP计划中总结经验形成案例向公众分享,因此FDA需要和申请方达成共识,哪些信息可以向公众披露,哪些信息须保密,在确定最终是否入选CDRP前,FDA会向申请方发送一份信息披露协议,说明哪些信息会被公布。
CDRP沟通途径(以CBER为例)
一般咨询:邮箱(industry.biologics@fda.hhs.gov)
IND相关沟通咨询:负责该IND的项目经理(推荐方式)
一旦这个IND 被纳入到了CDRP计划中,会议申请需要以amendment形式递交在该IND下
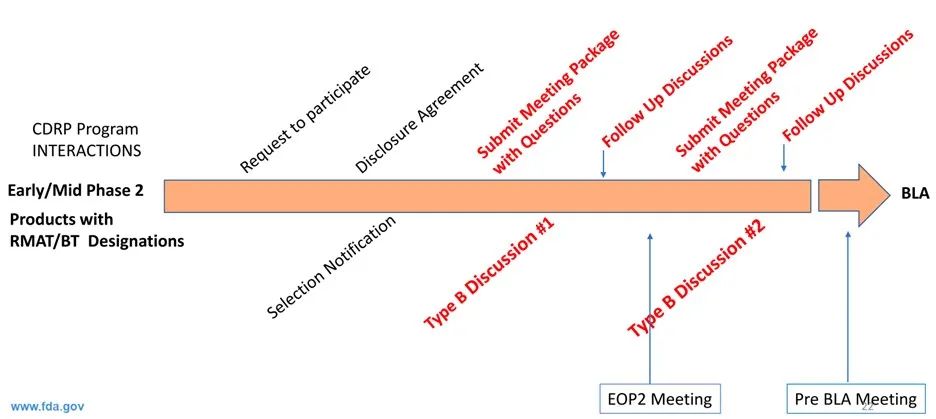
来源:CMC Development and Readiness Pilot (CDRP) Program, Regulatory Education for Industry (REdI)-2023 Annual Conference, June 8-9, 2023
其他要点
FDA计划每年批准9个IND纳入到CDRP计划中,其中CBER 6个,CDER 3个(CBER和CDER的数量会根据情况有所调整,但总数不变)
如果第一年CDRP被拒,申请者在第二年可以重新申请
FDA计划在2025年开展CDRP研讨会,与公众分享经验和案例,会后也会出台相关策略文件总结会议内容以及确定CDRP的后续发展
声明:本文内容部分源于FDA官网信息,中文翻译仅代表HPC观点,如有意见或想法,欢迎联系我们或留言,感谢。