本指南中规定的“物料(material)”包括:(1)试剂,滋养细胞(feeder cells),与起始物料、中间体、终产品直接接触的辅料(以及制剂中包含的其它非活性成分);(2)任何用于生产试剂、滋养细胞、辅料的物料;(3)引入TEMPs产品的物料。本指南中所指物料不包括:作为起始物料用于生产人细胞、组织、基于细胞/组织的产品(包括TEMPs)的人体细胞。
IND对人源/动物源物料的一般要求
• 信息和文件:以表格形式列出所有生产中使用的物料的基本信息,比如(包括但不限于)以下表格所列。这部分内容放在CTD的3.2.S.2.3(Control of Materials)和3.2.P.4 (Control of Excipients)模块。申报材料中递交相关证明文件,比如质量标准、检测报告(Certificates of Analysis, COA)、产地证明书(Certificates of Origin, COO)、包装说明书(package inserts)。
• 物料检测:对人源和动物源的物料,申报材料中还需要酌情递交物料来源和/或供应商对外来病毒因子检测的标准。如果供应商检测不足,那么生产的CGT或TEMPs产品可能还需要进行额外检测。对相关传染病病原体或疾病的检测,应使用FDA批准的检测试剂盒(21 CFR part 1271)。基于风险评估,申办方需要建立内部鉴别检测以防物料混淆。人源/动物源物料可能存在纯度、规格、质量等方面表现出的供体差异,因此批次之间质量可能不统一。申办方需要对可能会影响产品性能的属性建立质量标准,确保终产品质量保持一致。
• 物料等级:最好使用最高级别的物料,比如FDA批准的;或者,最好使用不含人源或动物蛋白的物料(比如使用不含人/动物源物料的组织培养基、重组蛋白)。
从人血和血液成分中提取的物料
人源物料通常指人血和血液成分中提取的物质,包括源血浆(Source Plasma),即专门用于制造血液制品的血浆,通过血浆分离术(plasmapheresis)从供者血液中分离出来。该定义不包括用于静脉注射的单供体血浆产品。用于生产CGT和TEMPs的物料可能来自于多种供体材料,比如全血、单供体血浆、源血浆都可以用来生产人AB血清(human AB serum)。但对源血浆的检测要求可能和对全血以及单供体血浆的检测要求不一样。比如源血浆仅仅用于进一步的生产制造的话,并不要求检测HTLV、WNV病毒和Chagas,而对供体梅毒的检测要求也有所不同。• 供体材料收集和检测:人血和血液成分的收集、处理、相容性测试、储存和分发必须按照现行良好生产规范的适用要求进行(21 CFR part 606)以及符合供者筛查和检测的要求(21 CFR part 630, subpart B, 21 CFR part 640, 21 CFR 610.40)。建议从FDA注册的机构购买。
• 降低TSE风险:克雅氏病(Creutzfeldt-Jakob Disease, CJD)和CJD变种疾病都是和输血感染相关的疾病,遵从2022年5月发布的CJD指导原则《Recommendations to Reduce the Possible Risk of Transmission of Creutzfeldt-Jakob Disease and Variant Creutzfeldt-Jakob Disease by Blood and Blood Components》。
• 常用的人源物料:
1. 血小板裂解物(Human Platelet Lysate, HPL):从破裂的血小板中分离出来的可溶性部分,常用作血清替代物加入到培养基中。
♢ 如果血小板是生产HPL的起始物料,申办方需要说明使用的血小板是否过期,因为血小板的生长因子稳定性可能受到储存时间的影响。对血小板建立接受标准(比如储存时间,一些生长因子的最低限度等);
♢ 血小板如何储存;
♢ 避免交叉污染,申办方需要描述HPL的制备过程,提供制备使用的材料设备信息,指出使用的设施。
2. 血清(human serum):通常从血浆、源血浆或全血中获得,比如AB血清。源血浆有特定的检测要求,因为它旨在用于进一步生产血浆来源的生物制品,这些生物制品使用经过验证的病毒灭活/去除工艺(如柱层析、洗涤剂处理或广泛的热灭活)生产。AB血清的生产通常没有这些工艺过程,因此源血浆作为起始物料生产AB血清并不合适,除非使用FDA批准的检测试剂盒检测HTLV,Chagas,WNV,babesiosis,梅毒等21 CFR 610.40规定的要求。记录AB血清整个工艺生产过程,包括供体材料收集、去纤维步骤(如有)、热灭活条件(时间和温度)、辐照(辐照类型和辐照剂量kGy)。
3. 人血白蛋白(human serum albumin, HSA):作为CGT或TEMP产品的辅料,使用的HSA必须是美国上市的产品。如果生产过程中使用但不是辅料,建议使用美国上市的或USP级别的HSA。如果使用的HSA不是美国上市的,需要提供使用理由;如果是美国上市的,需要提供产品说明书。
4. 培养基中的人源蛋白(human-derived proteins in culture media):人血蛋白可以加入培养基中,比如转铁蛋白是一种血液蛋白,通常作为补充剂添加到“无血清”、“低血清”、“无异种成分”或其他补充了特定成分的细胞培养基中。这些人源成分在物料COA中可能不会体现,因此申办方需要在文件中说明。同样,这些人源物料也要符合21 CFR 610.40的要求。如果供应商不方便提供详细信息,那么这些信息需要在CBER部门进行DMF备案,申办方递交IND时附上授权信(a letter of authorization, LoA)。
人源滋养细胞、旁观细胞(bystander cell)和细胞衍生颗粒(cell-derived particles)
在CGT和TEMPs产品的生产中,人源滋养细胞、旁观细胞和细胞衍生颗粒(比如胞外囊泡、外来体、分泌的蛋白)可以用来扩增细胞。比如永生化滋养细胞,经过高剂量辐射处理产生细胞颗粒的同种异体细胞,经过基因改造可以表达某些刺激蛋白的细胞。这类细胞的供体要求也要符合21 CFR part 1271, subpart C。对滋养细胞和旁观细胞的细胞库检测要求无菌、支原体、相关外源病毒因子,包括体内外源病毒因子检测(主细胞库),体外外源病毒因子检测(主细胞库和工作细胞库),TEM检测病毒颗粒,PCR检测人病原体,逆转录病毒。根据实际情况进行检测,如果细胞库样品检测有限,那么就要对原料药或产品制剂进行相关病毒检测。
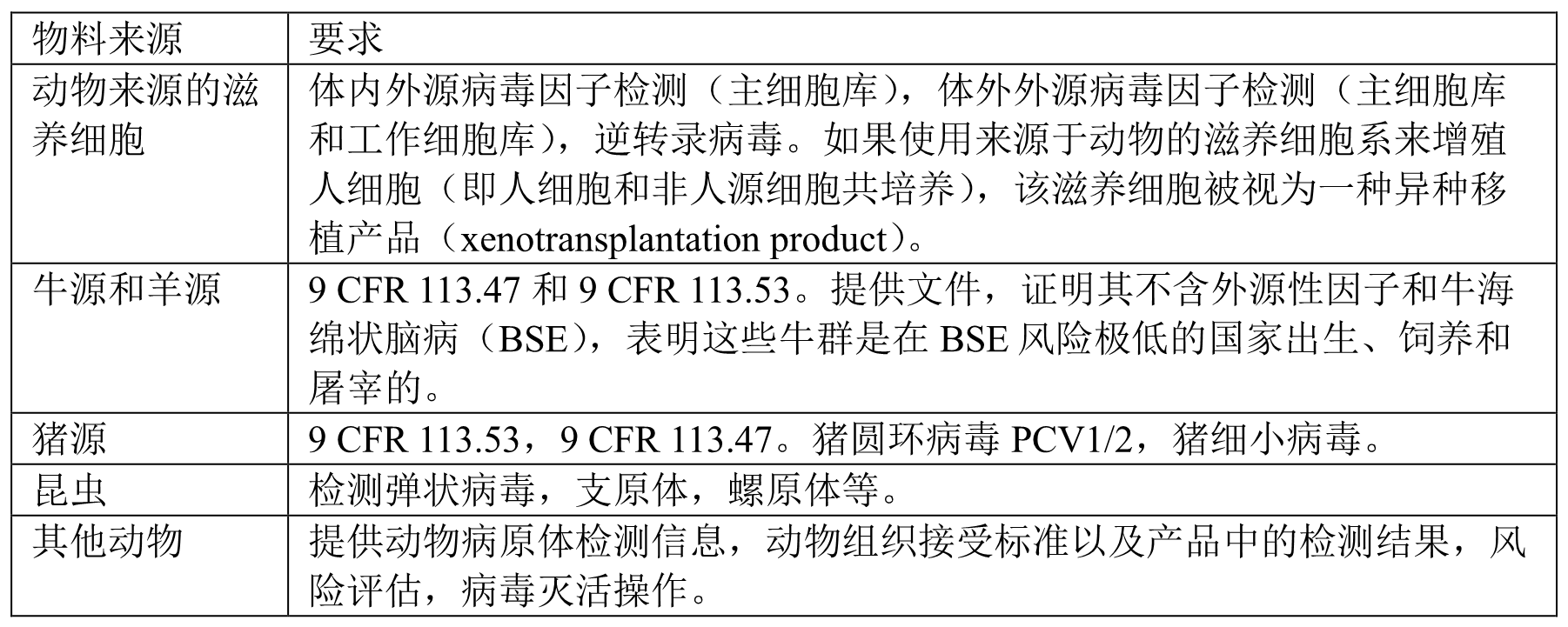
动物来源物料
所有动物源物料检测满足9 CFR 113.47 和 9 CFR 113.53的要求。